Editor's Choice:
Fluidigm develops a single-cell detection of miRNA expression profiles protocol on an automated manufacturing system and a single cell C1 TM Biomark TM HD system. This protocol allows 96 single cells to be processed in parallel in less than 24 hours, with up to 96 miRNAs per single cell in a single GE 96.96 chip. The SINGuLARTM 2.0 analysis software can perform unsupervised cluster analysis and PCA analysis on the data, effectively revealing the heterogeneity of the cell population at the single cell level according to the miRNA expression type, and provide more data for studying miRNA regulation.
Detection of MicroRNA Heterogeneity in Single Cells Using an Automated Microfluidic Chip System
Leyrat Anne, Shuga Joe, Li Nianzhen, Szpankowski Lukasz, Unger Marc & West Jay
(ISSCR 2013 poster: F-3201)
Introduction
MicroRNAs (miRNAs) are a class of short (18-24 nucleotide) non-coding RNAs that regulate gene expression by disrupting the stability of messenger RNA (mRNA) and inhibiting mRNA translation. Expression of miRNAs in cell populations is generally thought to drive downstream gene expression and protein function. Our goal was to determine changes in miRNA expression at the single cell level using a microfluidic system that automates single cell capture and miRNA preamplification for subsequent expression analysis. We developed a simple, modular process that can easily reduce the analysis of cell populations to single-cell levels (Figure 1). This process comprises two core components: C1 TM automated preparative single cell system (FIG. 1a: Preparation of a sample, comprising cells isolated and prepared from cDNA miRNA) chip and a dynamic (Dynamic Array ™ IFC) and BiomarkTM HD system (FIG. 1b: read , highly parallel expression analysis). Target Specific Amplification (STA) for each single cell captured by the C1 TM chip, using the Single Cell-to-CtTM kit (Life Technologies) to complete the lysis and pre-amplification steps, and TaqMan The ® MicroRNA Reverse Transcription Kit (Life Technologies) completes the reverse transcription step (Figure 2).
Using dynamic system Biomark TM HD chip and can use the expression of microRNA TaqMan 96 pairs of primers, parallel analysis of 96 cDNA samples obtained from preamplified 96 single cells. Principal Component Analysis (PCA) of the data using Fluidigm SINGuLARTM 2.0 analysis software revealed significant changes in miRNA expression in a population of single cells obtained from a single phenotype (Figures 3, 4, 5). Comparison of cell populations of different phenotypes (human embryonic fibroblasts, human induced pluripotent stem cells (iPS), human neural progenitor cells (NPC) obtained from iPS, and fully differentiated human neurons (HN)), except for the same type of cells In addition to the heterogeneity of expression, a greater difference (between different types of cells) is demonstrated.
result
Figure 1. Integration process for miRNA analysis in a single cell

The C1 single cell automated preparation system performs target-specific amplification (STA) of miRNA transcripts in single cells using reagents and experimental methods developed by Life Technologies ("Single-cell MicroRNA expression analysis"). The entire process from adding the cell suspension to the C1 chip to complete the data analysis can be completed in less than 24 hours.
Figure 2. C1 MicroRNA STA Experimental Procedure
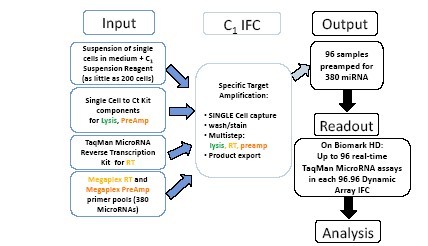
A single cell suspension (200-1000 cells) was added to the cell inlet of the C1 IFC chip. Lysis reagent from the pre-amplification and Single Cell-to-Ct ™ kit (Life Technologies), and reverse transcriptase reagents from TaqMan® MicroRNA Reverse Transcription kit (Life Technologies), and reverse transcription (using Megaplex TM RT pool ) and pre-amplification (using Megaplex TM PreAmp pool), so that each single cell can be detected up to 380 kinds of miRNA expression. C1 IFC captures single cells, then rinses, lyses, and reverse transcribes and preamplifies each single cell miRNA in parallel in the reaction chamber. 96 pre-amplified samples were then exported and run on a 96.96 dynamic chip using the Biomark HD system to study the expression of up to 96 miRNAs in each sample.
Figure 3. Analysis: Single iPS cells and their NPC descendants
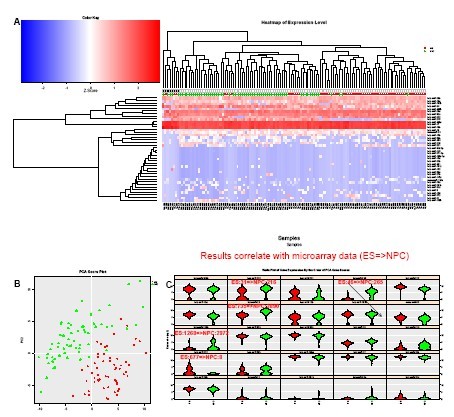
(A) Unsupervised cluster analysis of the data clearly distinguishes between iPS cells and NPC descendants obtained from iPS using small molecules. 1 Also reveals subpopulations in each cell.
(B) PCA clearly reveals the difference between the different cell types of the two phenotypes.
(C) Violin Plot shows differential expression of miRNAs in different subpopulations, as well as major contributors to principal components 1 and 2 (top left to bottom right order). The expression changes of the five miRNAs between iPS and NPC were the same as those obtained from embryonic stem cells (ES) and their NPC descendants using microassay (unpublished data, provided by Yao Shuyuan).
Figure 4. Human neurons, iPS and NPC cells
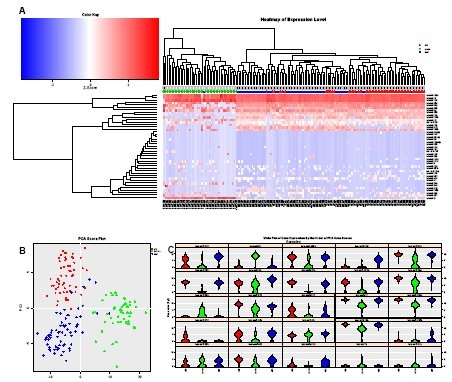
(A) Unsupervised clustering analysis of data obtained from iPS, NPC, and mature neurons (HN) clearly distinguishes HN cells from iPS and NPC cells, and also reveals the presence of each type of cell. Subgroup. miR-9 is expressed more frequently and at higher levels in mature neurons (HN).
(B) Based on miRNA expression, PCA can clearly distinguish these three types of cells. miR-20a, 19b, 17, and 106a are expressed at lower levels in HN, consistent with expectations based on neural differentiation and aging data 2,3 .
Figure 5. Embryonic fibroblasts with different passages
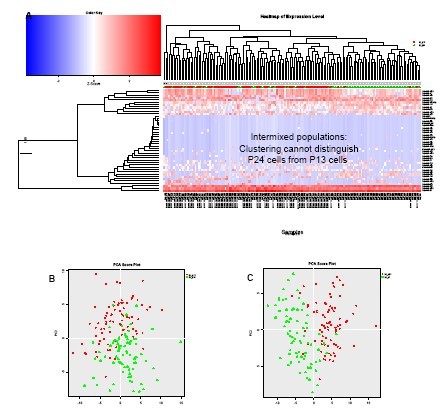
(A) Unsupervised clustering analysis of data from BJ embryonic fibroblasts obtained from two different passages (P13 and P24), although it can reveal differences in the types of miRNA expression between different cells, but it is impossible to distinguish between the two. Kind of group.
(B) PCA analysis of miRNA expression data obtained from P13 and P24 cells further confirmed that these two cells could not be distinguished based on miRNA expression.
(C) When the number of passages is further apart (P7 vs. P24), a PCA analysis of the miRNA data (heat map not shown) can distinguish them.
in conclusion
· We developed a simple protocol in the C1 TM single-cell automated preparation system that can process up to 96 single cells in parallel and analyze their miRNA expression profiles in less than 24 hours with minimal manual manipulation.
· The C1 miRNA STA protocol uses Life Technologies' miRNA-optimized reagents. In particular, Megaplex ™ RT and single cell PreAmp pool may each be up to 380 kinds of cDNA obtained from different miRNA C1 IFC. Using the Biomark system, the expression type can be read on the 96.96 GE dynamic chip.
• Unsupervised cluster analysis and PCA analysis of data from different cell types reveals differences in miRNA expression types between different cell types or in the same cell type (as confirmed by microarray or literature).
references
1. Chambers SM, et al. (2009) Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat Biotechnol. 27(3), 275-280.
2. Trompeter HI, Abbad H, Iwaniuk KM, Hafner M, Renwick N, et al. (2011) MicroRNAs MiR-17, MiR-20a, and MiR-106b Act in Concert to Modulate E2F Activity on Cell Cycle Arrest during Neuronal Lineage Differentiation of USSC . PLoS ONE 6(1): e16138. doi:10.1371/journal.pone.0016138
3. Hackl M., Brunner S., Fortschegger M., Laschober GT, Micutkova L., et al. (2010), miR-17, miR-19b, miR-20a, and miR-106a are down-regulated in human Aging . Aging Cell , 9(2), 291-296.
Detection of MicroRNA Heterogeneity in Single Cells Using an Automated
Introduction
Introduction
MicroRNA (miRNAs) are short (18–24 nucleotides), non-coding RNAs that regulate gene expression by both disrupting messenger RNA (mRNA) stability and inhibiting mRNA translation. The expression of miRNA species in cellular populations is thought to drive downstream gene expression And protein functionality. Our goal was to determine the variability in miRNA expression at the single cell level using a microfluidic system which automates single cell capture and miRNA pre-amplification for downstream expression analysis. We have developed a simple, modular workflow for streamlined analysis of Cell populations down to the single-cell level (Figure 1). The workflow is centered on two key components: the C1TM Single Cell Auto Prep System (Figure 1a: Sample Prep, including cell isolation and cDNA preparation from miRNA species) and the Dynamic ArrayTM IFC and BiomarkTM HD System (Figure 1b: Read out, for highly parallel expression analysis). The Specific Target Amplification (STA) chemistry Performed on each individual cell captured on the C1TM IFC borrows components from the Single Cell-to-CtTM kit (Life Technologies) for the lysis and preamplification steps and components from the TaqMan® MicroRNA Reverse Transcription Kit (Life Technologies) for the Reverse Transcription Step (Figure 2).
Using the Dynamic Array IFCs and the Biomark HD System, up to 96 cDNA samples preamplified from the 96 single cells are each analyzed in parallel with up to 96 microRNA TaqMan expression assays. Principal Component Analysis (PCA) of the data using Fluidigm's SINGuLARTM Analysis Toolset v2.0 reveals significant variations in the expression of discrete miRNA species in a population of single cells from a single phenotype (Figure 3, 4, and 5). Comparison of phenotypically distinct populations (human embryonic fibroblasts, human induced Pluripotent Stem Cells ( iPS), human Neural Progenitor Cells (NPC) derived from the iPS, and fully differentiated human neurons (HN)) demonstrate more dramatic differences in addition to the heterogeneity of expression within each group.
Results
Figure 1: Integrated workflow for miRNA analysis in single cells
The C1 Single-Cell Auto Prep System performs Specific Target Amplification (STA) of miRNA transcripts from single cells using the reagents and a protocol developed for this purpose by Life Technologies (protocol "Single-cell MicroRNA expression analysis"). The whole process, From loading the cell suspension on the C1 Integrated Fluidic Circuit (IFC) to full data analysis of the data can be accomplished in less than 24 hours.
Figure 2. C1 MicroRNA STA original workflow
Figure 3. Analysis: Single iPS cells and their NPC progeny
A) Unsupervised clustering of the data clearly distinguishes iPS cells from their NPC progeny obtained using small molecules1. Subpopulations are also revealed within each group of cells. B) PCA shows a clear difference between the two phenotypically distinct cell populations. C) Violin plots show Differential variations of miRNAs in different subpopulations and reveal the main contributors to Principal Components 1 and 2 (in order from top left to right). The variations in expression of a set of five miRNAs between iPS and NPC shows the same trends as microarray measurements obtained With Embryonic Stem cells (ES) and their NPC progeny (unpublished data, courtesy of Yao Shuyuan).
Figure 4. Human Neurons, iPS and NPC cells
A) Unsupervised clustering of the data obtained with iPS, NPC and mature neurons (HN) clearly distinguishes HN cells from iPS and NPC cells and also reveals subpopulations within each cell type. miR-9 is more frequently and more highly expressed in mature neurons ( HN). B) PCA clearly distinguishes between the three cell types based on miRNA expression. The expression of miR-20a, 19b, 17 & 106a is lower in HN, as expected based on neural differentiation and aging data2,3 .
Figure 5. Embryonic fibroblasts at different passage number
A) Unsupervised clustering of the data from two different cultures of BJ embryonic fibroblasts obtained at difference passage numbers (P13 and P24) is not able to distinguish the populations from one another, even though it can reveal different miRNA expression patterns between individual cells. B PCA analysis of the miRNA expression data from P13 and P24 cells confirms that the two cell populations are undistinguishable based on miRNA expression. C) When the passage numbers are more distant (P7 vs. P24), PCA analysis of the miRNA data (heatmap) Not shown) distinguishes passage number P7 from P24.
Conclusion
• We have developed a streamlined protocol on the C1TSingle-Cell Auto Prep System to analyze the expression patterns of miRNA species in up to 96 individual cells processed in parallel with minimum hands-on time, in less than 24 hours.
A) Unsupervised clustering of the data from two different cultures of BJ embryonic fibroblasts obtained at difference passage numbers (P13 and P24) is not able to distinguish the populations from one another, even though it can reveal different miRNA expression patterns between individual cells. B PCA analysis of the miRNA expression data from P13 and P24 cells confirms that the two cell populations are undistinguishable based on miRNA expression. C) When the passage numbers are more distant (P7 vs. P24), PCA analysis of the miRNA data (heatmap) Not shown) distinguishes passage number P7 from P24.
Conclusion
• We have developed a streamlined protocol on the C1TSingle-Cell Auto Prep System to analyze the expression patterns of miRNA species in up to 96 individual cells processed in parallel with minimum hands-on time, in less than 24 hours.
• The C1 miRNA STA protocol uses reagents optimized by Life Technologies for miRNA analysis. In particular, the Megaplex ™ pools of RT and PreAmp primers allow to produce cDNA from up to 380 different miRNA species in each cell processed in the C1 IFC. The expression Patterns are read out using the Biomark HD System on 96.96 GE Dynamic Array IFCs.
• Unsupervised clustering analysis and PCA of the miRNA data from different cell types reveal different patterns of miRNA expression between the different cell types (confirmed by microarray data or the literature) and also within each cell type.
Surgical Drapes Disposable,Fenestrated Surgical Drape,3 Ply Surgical Drape,2 Ply Surgical Drape
Suzhou JaneE Medical Technology Co., Ltd. , https://www.janeemedical.com